液相色谱法中应该怎么解释组分的洗脱顺序?
 popxyp0202022-10-04 11:39:542条回答
popxyp0202022-10-04 11:39:542条回答
已提交,审核后显示!提交回复
共2条回复
 谢彤 共回答了23个问题
谢彤 共回答了23个问题 |采纳率82.6%- 根据你样品成分的极性来解释,同时还要看你使用的正向柱还是反相柱,如果用的是反向色谱柱(C8、C18等),色谱柱填料极性较弱,那么被分析的物质就是极性大的先出峰;如果用的是正向色谱柱(CN、NH2),色谱柱填料极性较强,那么被分析的物质就是极性小的先出峰.
- 1年前
- wsjfff 共回答了19个问题
|采纳率 - 根据极性来解释,大的后出峰。
- 1年前
相关推荐
- 液相色谱法中峰高为多少样品可进行二级稀释
傻蹲街边1年前1
-
yueyu8161 共回答了15个问题
|采纳率86.7%其实主要关注的不是峰高,而是柱子是否过载.当然,正常的峰高度最好不要超过仪器最大量程的2/3.
当峰平顶,峰形前延且不尖锐时,通常会考虑是否柱子因为样品浓度高而过载,可以尝试二次稀释后重新分析.
或者如果你用外标法定量时,峰面积超过标准品最高浓度对应的面积时,必须进行稀释,使得样品浓度在外标工作曲线范围之内.1年前查看全部
- 用高效液相色谱法进行药物鉴别的问题,
用高效液相色谱法进行药物鉴别的问题,
某复方制剂中非那西丁鉴别方法的建立
用十八烷基硅烷键合硅胶为填充剂,以甲醇-水(70:30)为流动相,检测波长为254nm,理论板数按非那西丁峰计算不低于1500,非那西丁峰和内标物质福美斯坦峰的分离度应符合要求.取含量测定项下的溶液进行进样测定,供试品溶液的主峰保留时间应与非那西丁对照品的保留时间一致.测定结果见图
非那西丁的高效液相色谱
A:对照品;B:供试品;C:空白样品
1.内标物峰;2.非那西丁峰
用高效液相色谱法进行药物鉴别时,方法应如何设计?在本例中图A、图B和图C各说明了什么问题?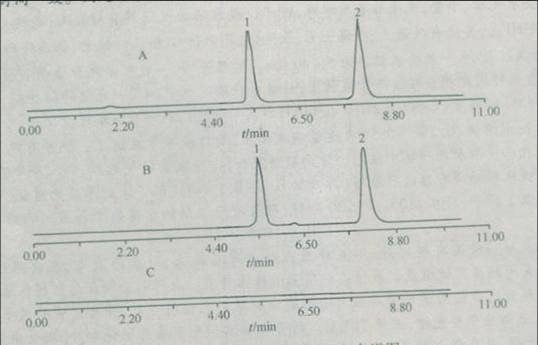 delpiero8301年前1
delpiero8301年前1 -
梨子7 共回答了11个问题
|采纳率90.9%原则:选用保留时间或相对保留时间作为鉴别主要内容
实验设计的考察因素:
1、色谱条件适用性考察,一个复杂的过程,分别对检测波长、色谱柱、流动相(PH、配比)、流速、温度等因素进行确定,已达到最适宜的分离度、对称度、重复性、理论板数等色谱关键因素,加入内标物可的减少重复性造成的误差,根据相对保留时间进行鉴别,对于是否需加入内标物,及内标物的选取还要进行考察;
2、各因素考察,关键性内容,一个庞大的实验过程,分别对准确度(次要)、精密度(次要)、检出限(次要)、专属性(主要)等因素进行试验,用以确定试验的适用性及有效性;
3、做好以上工作基本可以确立初期的试验方法,经过长期试验考察后(上述内容的回归分析)可能会发现其中的不适用部分,仍需要对方法进行完善,从而得到较成熟的方法,此项工作属于不可终止工作,一直处于完善过程.
以上只是简单的概述,具体的各项内容可以参照相关范本.
A图:可以根据此图计算对于内标物的相对保留时间,作为鉴别依据指标;2分钟处有一小峰,由B、C图可判定此杂质为对照品中杂质,不是内标物引入,杂质峰对主峰无干扰可进行鉴别判定.
B图:可计算样品的相对保留时间,与上边结果比对,用以结果判定;在6.3分钟左右有一杂质峰,根据A、B图可判定为样品引入;杂质峰对主峰保留时间基本无干扰可以进行鉴别判定.
C图:A,B图的前提,表明空白溶剂对试验是否存在干扰,此图表明空白溶剂无干扰.1年前查看全部
- 液相色谱法用外标法检测含量时两个平行试验相差大!
液相色谱法用外标法检测含量时两个平行试验相差大!
我做的单一成分,cn1217181年前1 -
1121114 共回答了16个问题
|采纳率100%外标法只看峰面积而不看其他成分,因此平行试验间相差过大的话都是实际进样量产生了差异,这可能的因素有:
定量环实际进样量出现差别,可能是定子密封圈擦划造成渗漏形成,或者是进样量未能充分冲洗并充满定量环;
进样浓度出现差别,可能是配制样品溶液时称量出现误差或者转移样品时出现溅撒等现象;
最后还可能是氘灯能量不足使检测不稳定或者泵内出现微小气泡使实际进柱子样品量变化等系统稳定性的原因,不过此类问题应较为明显,易发现.1年前查看全部
- 对于高效液相色谱法测定中使用标准曲线法的优缺点?
多情的梦梦1年前1
-
大春和小春 共回答了20个问题
|采纳率90%优点:线性稳定、良好、准确.
缺点:耗费时间很长.1年前查看全部
- 为什么硝基苯浓度的测定标准中没有高效液相色谱法,而只有气相色谱法呢?
为什么硝基苯浓度的测定标准中没有高效液相色谱法,而只有气相色谱法呢?
其实高效液相色谱法好像也可以测水中硝基苯的浓度,因为有很多相关的文献,但是为什么标准中选择的是气相色谱法呢?或者哪个标准是关于用高效液相色谱法测定硝基苯浓度的?hhy026a1年前3 -
uuyhc 共回答了21个问题
|采纳率76.2%一般而言,能选用气相色谱方法的就尽量选择气相色谱方法,气相定量比较困难才考虑液相色谱方法.因为气相操作比较简单,消耗品的使用寿命及维护比液相要小一些.“色谱世界”网站的色谱图库有硝基苯的色谱图,你去对比看看就知道了.1年前查看全部
- 液相联质谱法和液相色谱法什么区别?
液相联质谱法和液相色谱法什么区别?
液相还分哪几种啊,我只知道有很多不同的检测器,请懂得回答的详细点,blackteaxu1年前2 -
恋你的单飞鹰 共回答了14个问题
|采纳率100%HPLC从不同的角度出发可以有不同的分类方法,一般液相色谱根据柱子填料和流动相选择的不同分为正相色谱和反相色谱.所说的“高效”和“超高效”,则是涵盖了小颗粒填料、非常低系统体积及快速检测手段等技术.
一般的HPLC都会配有检测器,液相色谱/质谱联用,是在HPLC后再加一个MS(质谱)检测器.
补充两点:
1,由于HPLC检测器(常见为UV类)和MS工作原理不同,有些化合物不会同时在UV和MS上有响应.
2,由于MS检测器对于流动相有要求,比如易挥发,低盐等,有时HPLC适合的流动相条件并不能直接用在液/质联用仪上,需作改动.1年前查看全部
- 请问用高效液相色谱法测定植物激素,测得结果是内源激素还是激素总量呢?
请问用高效液相色谱法测定植物激素,测得结果是内源激素还是激素总量呢?
HPLC法测牡丹花芽内赤霉素含量cocoon5121年前1 -
wuxingzhe123 共回答了22个问题
|采纳率90.9%植物内源激素的反相高效液相色谱法测定--《分析测试学报》2001年01期1年前查看全部
- 用高效液相色谱法做一个药品中某种成分含量的测定需要多长时间?
kopeok1年前1
-
办良心事 共回答了18个问题
|采纳率100%每针大概需要10分钟左右吧,要看你需要做几组试验了,其实实验过程很短的,关键是实验前准备过程较长,高效液相色谱仪的仪器清洗大概需要20个小时左右吧,当然如果上一次做试验的人员已经把仪器清洗好了就不会花费很长时间了!1年前查看全部
- 用已建立的高效液相色谱法(色谱条件略)测定某中成药粒剂栀子苷的含量,对照品栀子苷的称量为10.01MG,用甲醇溶解定容至
用已建立的高效液相色谱法(色谱条件略)测定某中成药粒剂栀子苷的含量,对照品栀子苷的称量为10.01MG,用甲醇溶解定容至100ML,连续10微升,进行三针的锋面积A分别是409、411、421.样品研细后分别称样0.1002G(1号)和0.1013(2号)分别置于25ML量瓶中,各用22ML甲醇溶液,超声30分钟后用甲醇稀释至刻度,混匀,取上清高速离心后进样10微升分析,发现峰面积远大于对照品溶液峰面积分别为387、389(1号)和398、401(2号)请计算中成药颗粒的栀子苷含量(单位MG/G)159412356991年前2
-
rainbowzhengrong 共回答了21个问题
|采纳率95.2%用已建立的高效液相色谱法(色谱条件略)测定某中成药粒剂栀子苷的含量,对照品栀子苷的称量为10.01MG,用甲醇溶解定容至100ML,连续10微升,进行三针的锋面积A分别是409、411、421.样品研细后分别称样0.1002G(1号)和0.1013(2号)分别置于25ML量瓶中,各用22ML甲醇溶液,超声30分钟后用甲醇稀释至刻度,混匀,取上清高速离心后进样10微升分析,发现峰面积远大于对照品溶液峰面积分别为387、389(1号)和398、401(2号)请计算中成药颗粒的栀子苷含量(单位MG/G)1年前查看全部
- 在高效液相色谱法中,与液体固相相比较,化学键合固定相有何优点
在线只为清清草1年前1
-
zy222szy 共回答了27个问题
|采纳率88.9%化学键合固定相的有点:
1使用过程不流失
2化学性能稳定
3热稳定性好
4载样量大
5适宜做梯度洗脱1年前查看全部
- 液相色谱法,吸附色谱中的吸附剂和洗脱剂
液相色谱法,吸附色谱中的吸附剂和洗脱剂
吸附剂是不是固定在固定相中的?洗脱剂是指液相,还是指液相以外加入的其他溶剂?洗脱剂的作用又是什么?ztejyhtao1年前2 -
g_sniper 共回答了15个问题
|采纳率93.3%首先吸附剂是固定在固定相中的 洗脱剂为所用的流动相 作用是携带待测组分在色谱柱内向前移动并流出色谱柱1年前查看全部
- 提取物中含有双黄酮,用高效液相色谱法检测其中双黄酮含量.为什么能检测出来?而不是总黄酮含量
提取物中含有双黄酮,用高效液相色谱法检测其中双黄酮含量.为什么能检测出来?而不是总黄酮含量
超声提取后的提取液中含有双黄酮,也含有其他的黄酮类化合物.为什么检测的就是双黄酮的含量,其他的黄酮有什么影响?无情ii人1年前1 -
sammbe 共回答了25个问题
|采纳率80%高效液相色谱本身也是一个分离纯化的仪器.原理是不同物质与高效液相色谱柱的结合能力不同,然后被液相洗脱的时间不同而达到分离的效果.换而言之,在进行高效液相色谱检测的时候,黄酮类物质会因为不同的洗脱时间而被分离开来,所以在条件优化的比较理想的情况下,你所检测到的就是你要的双黄酮,而其他黄酮类物质因为在柱子中的滞留时间与双黄酮不同,而先洗脱出或者后洗脱出来,所以不会影响到双黄酮的检测.1年前查看全部
- 如何用高效液相色谱法测定未知样品
lianlianqingjie1年前1
-
ufang 共回答了20个问题
|采纳率90%高效液相色谱,其实就是一个将样品中复杂成分进行分离处理的工具,能不能分离,分离的好不好,还受制样水平,使用的柱子和溶剂合理不合理,等方面的限制.即使你都分离好了,不通过核磁、质谱、红外、紫外等其他仪器的帮助,也不能确认你的未知样到底是什么.1年前查看全部
- 气相色谱法和液相色谱法的相同点和不同点.
鬼影狂刀1年前1
-
yefeng520060 共回答了17个问题
|采纳率82.4%这个问题有点太大了,我只能挑重点说一两个.相同点:它们都是色谱法,也就是利用不同物质在不同相态的选择性分配,以流动相对固定相中的混合物进行洗脱,混合物中不同的物质会以不同的速度沿固定相移动,最终达到分离的效果.简单一点说,它们的原理都是一样的.不同点:它们检测的物质不同.气相色谱法,顾名思义主要是检测气体.当然还有很多沸点比较低的液体.然而液相色谱法则是检查溶液状态的样品,比如液体或者配制成溶液的固体.尤其是液相色谱需要检查有紫外吸收的物质.流动相不同:其实所谓的液相色谱和气相色谱主要的名称来源就是他们的流动相.液相色谱的流动相是液体,而气相色谱的流动相是载气.1年前查看全部
- 如何建立高效液相色谱法测定含量的方法
佑元1年前1
-
lhy916 共回答了19个问题
|采纳率84.2%1 色谱条件的确定
专属性是色谱条件建立的关键通常是采用
在被测物对照品(或供试品)中加入适量的杂质或辅
料以验证所选色谱条件能否将各杂质与被测物
分离检出
[2]
.应按1(w/w)被测物浓度的各杂质量
添加至被测物中模拟被测物中可能存在杂质的
状态即有少量(约1)杂质存在时能否与被测物
达到完全分离(分离度大于1.5)以验证系统适用
性.只有这样才能较为客观、科学地反映被测物的
实际情况.而不应将被测物与各杂质配制成相同浓
度的溶液因为实际检测中不可能存在这种情况
且该浓度也不易确定.在实际检测时由于被测物
浓度较大很易将相邻杂质峰包含其中.另外还需
测定溶剂和辅料(检测制剂时)是否有干扰.目前
美国药典(USP)、英国药典(BP)及许多进口产品的
质量标准中有关物质测定方法学的专属性验证均
采用此法.还须说明的是杂质与杂质峰间的分离
度达1.2即可而被测物与其相邻杂质峰的分离度
必须大于1.5.
2 检测波长的选择
有关物质检测的研究对象是杂质而非被测
物.但测定则是通过各自的峰面积来表达故波长
的选择必须考虑被测物和各杂质在检测波长下的校
正因子(f)是否相同.应分别制备相同浓度的被测
物与各杂质溶液经紫外扫描后以吸光度相近的波
长为检测波长.在该检测波长下分别进样测定
由各峰面积计算校正因子.若f为0.81.2则表明
被测物与各杂质的f相同可消除f的影响.若f≤0.8
或f ≥1.2则应在计算时加入f.目前通常以被测物
的最大吸收波长为检测波长、不加校正因子的计算
方法而未综合考虑各杂质的f.
3 供试品溶液浓度的确定
供试品溶液浓度的确定也非常重要.虽然浓度
越高越能反映被测物中杂质存在的情况但若设定
过高会产生主峰严重拖尾、裂峰、柱超载和检测器超载等情况若设定过低则灵敏度不够无法
检测杂质及其含量变化.
最低检出浓度的测定可分为信噪比法和直接评
价法两种
[3]
.后法是目前较为科学的做法即将仪
器的灵敏度调至较适宜的值(仅对灵敏度可调节的
仪器而言目前市场上主流品牌的液相色谱仪均已
设定了一个恒定、较为灵敏的值)然后将被测物
溶液不断稀释后进样测定直至被测物峰面积无法
检出为止此时的浓度即为最低检出浓度.
最大进样量则是采用不断增加被测物溶液浓
度直至峰严重拖尾、裂峰、柱超载和检测器超载
等情况出现.
根据最低检出浓度采用“上推法”来确定
供试品溶液浓度如一般设定杂质总量小于1.0
对照液对照溶液的浓度至少应为最低检出浓度的
2050倍供试品溶液浓度则应是最低检出浓度的
20005000倍.同时还应考虑仪器、色谱柱等因
素对最低检出浓度和最大进样浓度的影响(即耐用
性因素)所以供试品溶液的浓度应在保证小于最
大进样量的情况下适当设定得高些以保证该浓
度在任何试验条件下均有足够的检测灵敏度.表
1为最低检出浓度、最大进样量、供试品溶液和对
照溶液间的比例关系(进样量10µl规定杂质限度
1.0).
4 线性试验
在稳定性考察中如某杂质含量不断增加
则说明被测物降解的途径稳定、可循则有必要对
该杂质进行针对性地监控即采用该杂质对照品
(经确证结构后由人工合成获得)以外标法准确测
定.此时与含量测定相似应进行线性试验.
通常将杂质限度设定为该杂质的100浓度线性
验证范围10150(即相当于被测物测定浓度
表1 最低检出量、最大进样量、供试品溶液和对照
溶液间的比例关系
参数浓度/µg·ml
–
1
绝对量/ng相当于供试品
溶液浓度/与最低检出
浓度的倍数
最大进样量3000
最低检出浓度0.110.02
供1年前查看全部
- 哪里有很好的高效液相色谱法的很好的理论
rain3231年前1
-
huohuliulanqi 共回答了17个问题
|采纳率88.2%说了这么多的很好,看来是想要很好的资料了.建议你如果看液相分析方面的理论就选择《药物分析》是安登魁的,还有一本叫《实用高效液相色谱分析方法建立》这本书也非常不错(这是来翻译国外的一本书,英文版叫《Practical HPLC Method Development 》).
这两本书都不错,从理论上讲的非常详细,明了.
基于一些基本理论,好多书上说的都有一定差异,所以理论上建议你看这两本书,而涉及的一些专业术语,要以中国药典2010为准.
这样学下来的基本理论扎实,学好之后就是正规军.不要乱看其它的书,浪费时间,进步还慢.
如果想研究仪器方面的建议看安捷伦消耗品手册,2012版的,那叫两个字:专业.1年前查看全部
- 高效液相色谱法 .执业药师考题高效液相色谱法常用于进行鉴别的参数是A、保留值B、峰高C、半高峰宽D、理论板数E、比移植我
高效液相色谱法 .执业药师考题
高效液相色谱法常用于进行鉴别的参数是
A、保留值
B、峰高
C、半高峰宽
D、理论板数
E、比移植
我实在是弄不懂,为啥不是D.....执业药师考题林建岳1年前2 -
bin_song 共回答了17个问题
|采纳率88.2%我记得鉴别用的是保留时间,不同的物质保留时间不同,相同的物质保留时间相同,因此可以用来鉴别.理论板数影响的是区分度,理论板数越大,能区分开的多种物质的峰就分离的越明显,理论板数小,物质峰可能会有重叠.因此从理论板数无法看出是哪种物质.1年前查看全部
- 高效液相色谱法的测定高效液相色谱法测定样品中巴比妥钠含量1.实验目的是什么?2.实验条件,室内温度及相对温度是什么?3.
高效液相色谱法的测定
高效液相色谱法测定样品中巴比妥钠含量
1.实验目的是什么?
2.实验条件,室内温度及相对温度是什么?
3.样品的准备,样品的取用.
4.仪器操作
5.数据处理 定量法的说明dfg212fh1年前1 -
寻宝大侠 共回答了11个问题
|采纳率90.9%巴比妥钠是巴比妥酸的钠盐,没有生理活性,我想你问的应该是苯巴比妥钠或异戊巴比妥钠等,在5位次甲基上有取代的才有生理活性.
你的问题象是个实验报告,悬赏分虽然很高,但直接给你一篇是没有意义的,应该把涉及的问题搞清楚.
1.实验目的 巴比妥类药物的含量测定一般采用经典的容量分析法——银量法、溴量法、酸碱滴定等,还有UV.HPLC多用于制剂及血液和尿液中巴比妥类药物的含量测定,为什么呢?因为需要先分离,后定量.
2.实验条件 你所说的室内温度及相对温度,没有严格的要求,仪器本身对试验环境是有要求的,但在室内一般都可以满足.
3.样品的准备,样品的取用 这个比较复杂.首先是配制对照品溶液(比较简单),还可以根据需要选用内标;其次是样品处理,以血药浓度测定为例,先要分出血清,再用有机溶剂萃取,在控温水浴上用氮气吹干,用流动相转移定容.样品处理比较简单的是用固相萃取,但成本比较高;还可以用柱切换在线分离,但对仪器硬件要求较高.
4.仪器操作 这个就不是几句话能说清楚的了,需要学习培训.色谱条件倒是可以从文献中查到,比如苯巴比妥钠的测定,C18柱,ACN-Water(60:40),波长254nm.
5.数据处理 定量法的说明 定量法不外乎外标法和内标法,一般都可以用一点法,如果线性不好,或截距比较大,可以用两点法或标准曲线.你应该在方法学的研究中确定.
不知道你的专业程度如何,我说的也许太浅显或太深了,希望对你有点帮助.1年前查看全部
- 结晶紫系列物质可否用同种液相色谱法分析 能有结晶紫内酯的液相色谱分析更好
4133071331年前1
-
新月灵澈 共回答了18个问题
|采纳率94.4%结晶紫 结晶紫是碱性染料,能溶于水(溶解度9%)和酒精(溶解度8.75%).
液相色谱所用的色谱柱是不能直接分离碱性物质的,会对硅胶基质产生永久性损害.
另外,结晶紫上有氯离子,氯对一般的色谱柱也有损害.
从结晶紫的结构上看,在紫外区是有较强吸收的,紫外检测器可用.
色谱柱的选择是您首要的问题,其次还有对结晶紫物质的PH调节也是您要考虑的问题.1年前查看全部
大家在问
- 1已知:如图,点B是⊙O外的一点,以B为顶点的角的两边分别交⊙O于点A、D和点C、E,BO平分角ABC.求证:AD=CE.
- 2once,twice,thrice后面是什么?
- 3要写过程的哈封冻的冰面上,一个体重为600牛的人,他的每只脚的鞋底面积为125平方厘米时恰好能走过冰面.如果另一个体重相
- 4英语中未来将发生的事情 going to do 和will有什么不同 怎么区分 区别在哪里?
- 5理科高手帮忙,设f(x-1/x)=x^2/(1+x^4),求f(x),希望给出解的过程.谢谢
- 6y=-2x平方函数图像咋画
- 7以成语为谜语猜汉字1.表里如一 谜底:_____2.顶天立地 谜底:_____3.以身作则 谜底:_____4.巧夺天工
- 8‘整个冬天’用英文说(除了all the winter)
- 9试画图表示前角=10°、后角=6°、主偏角=90°、副偏角=10°、刃倾角=-5°的外圆车刀
- 10根据已知地点纬度,计算日照角度,如北纬30度,冬至那天日照高度?
- 11如图,△ABC内接于⊙O,AB为⊙O的直径,∠BAC=2∠B,AC=6,过点A作⊙O的切线与OC的延长线交于点P,求PA
- 12补写出下列名篇名句的空缺部分。(1)今宵酒醒何处,
- 13《国际贸易实务》多项选择题 下列描述正确的是()
- 14两根同样长的绳子,第一根剪去它的一半,第二根剪去0.5米,剩下的两段绳子( )
- 15开头是two boys live in different places.they don't