测序的分辨率(resolution)是什么意思?在高通量测序里经常出现的词汇
 罪过啊2022-10-04 11:39:542条回答
罪过啊2022-10-04 11:39:542条回答
已提交,审核后显示!提交回复
共2条回复
 QQGAMW 共回答了15个问题
QQGAMW 共回答了15个问题 |采纳率80%- esolution 在测序的时候是指对碱基信号的分辨能力,尤其是在达到测序极限后(一般的机器为几百碱基对,就会出现信号模糊,无法再分辨了,这就是分辨率不行了
- 1年前
- lxl_l 共回答了16个问题
|采纳率 - 问题补充:还有一个相位的概念,是什么意思啊?能形象的解释吗? Here, we是这样的,高通量测序分为好多种,有不同的平台运作,共同的特点是极高的
- 1年前
相关推荐
- 关于illumina测序,basespace是什么意思 base这里是碱基的意思么?懂生物信息学和高通量测序的帮帮忙
关于illumina测序,basespace是什么意思 base这里是碱基的意思么?懂生物信息学和高通量测序的帮帮忙
关于illumina测序,basespace是什么意思base这里是碱基的意思么?懂生物信息学和高通量测序的帮帮忙 谢谢了...
 东快uu达人1年前1
东快uu达人1年前1 -
cjkg 共回答了22个问题
|采纳率81.8%BaseSpace is Illumina’s genomics cloud computing environment for next-generation sequencing (NGS) data analysis.Now biologists and informaticians can easily and securely analyze,archive,and share sequencing data.NGS data analysis is simplified and accelerated with push-button tools.Cumbersome and time-consuming data transfer steps are eliminated.Productivity is improved.摘自illumina官网.
BaseSpace就是云计算环境了.具体你去官网查查吧!竟然在百度问这么专业的问题,厉害!1年前查看全部
- 转录组测序和数字表达谱测序有什么区别
lina78330531年前1
-
fgtregtt 共回答了17个问题
|采纳率88.2%转录组测序和数字表达谱测序相比,主要有如下不同:x0d第一,测序目标不同.x0d转录组测序可以测定特定组织中全部mRNA,而表达谱测序只是测定mRNA的酶切标签序列(21 bp);x0d第二,代表性不同.x0d数字表达谱测序只测定21bp序列,而转录组测序测定转录本全长,因而可以更准确地代表样品转录表达情况;x0d第三,应用范围不同.x0d转录组测序应用范围广泛,不仅可以检测表达量差异,而且可以发现新的转录本和可变剪切等.而表达谱测序只能粗略检测表达量差异,不能反映基因转录表达的特点和规律;x0d第四,参考序列要求不同.x0d转录组测序不仅可以适用于基因组序列已知的物种,而且也适用于基因组序列未知的物种.而表达谱测序只适用于基因组序列已知的物种.1年前查看全部
- 对果蝇基因组进行研究时,应对几条染色体组进行测序
cqyjy1年前1
-
fengjiyue 共回答了12个问题
|采纳率91.7%果蝇是二倍体,它的染色体数目是4对,6 + XY.
所以,在进行基因组研究时,只需测定3 + XY ,即 5 条染色体即可.1年前查看全部
- 果蝇是非常小的蝇类,如图是科学家对果蝇正常染色体上部分基因的测序结果.请据图回答:
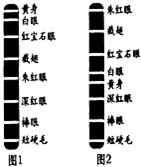 果蝇是非常小的蝇类,如图是科学家对果蝇正常染色体上部分基因的测序结果.请据图回答:
果蝇是非常小的蝇类,如图是科学家对果蝇正常染色体上部分基因的测序结果.请据图回答:
(1)根据图1,控制黄身和朱红眼的两个基因在形成配子时,______(能或不能)遵循基因自由组合定律.其原因是______.
(2)果蝇体内的图1染色体上所呈现的基因,不一定能在后代中全部表达,可能的原因是:______.(答出一点即可)
(3)与图1相比,图2发生的变异是______.秋秋QQ1年前1 -
anjingg 共回答了18个问题
|采纳率100%解题思路:基因自由组合定律的实质是位于非同源染色体上的非等位基因的分离或组合是互不干扰的,在减数分裂过程中,同源染色体上的等位基因彼此分离的同时,非同源染色体上的非等位基因自由组合.排列在染色体上的基因的数目或排列顺序发生改变,而导致性状的变异为染色体结构变异.染色体中某一片段位置颠倒180°后重新结合到原部位引起的变异是倒位.(1)基因自由组合定律的实质是减数分裂形成配子时,位于非同源染色体上的非等位基因自由组合随配子遗传给后代.而控制黄身和朱红眼的两个基因位于同一条染色体上(不是位于非同源染色体上).
(2)影响基因表达的因素既有内因,又有外因,如杂合子中的隐性基因不能表达、基因的选择性表达、以及基因的表达与环境有关等.
(3)对比图1与图2中的基因排序可以看出:图1中的黄身、白眼与、宝石眼等五个基因的顺序在图2中发生了180°的颠倒,这种变异属于染色体结构的变异.
故答:
(1)不能控制黄色和朱红眼的两个基因位于同一条染色体上(不是位于非同源染色体上)
(2)①当出现杂合子时,隐性基因不能表达②基因的选择性表达
③基因的表达与环境有关(答出任意一项均给分)
(3)染色体结构变异(染色体片段倒置或倒位)点评:
本题考点: 基因的自由组合规律的实质及应用;细胞的分化;染色体结构变异的基本类型.
考点点评: 本题考查果蝇基因与染色体关系的相关知识,意在考查学生的识记能力和判断能力,运用所学知识综合分析问题的能力,本题难度中等,属于考纲理解层次.解答本题的关键是理解基因与染色体的关系以及等位基因的概念.1年前查看全部
- 基因重组中碱基突变的原因?先用T载体将目的基因进行了连接,测序之后是正确的.但后用PET28a构建,测序发现丢失一个碱基
基因重组中碱基突变的原因?
先用T载体将目的基因进行了连接,测序之后是正确的.但后用PET28a构建,测序发现丢失一个碱基,并有一个碱基突变.会有什么原因?
构建中什么原因会导致突变,如何避免?anqierhjl1年前1 -
mbp1975 共回答了21个问题
|采纳率81%一般来讲有三种
物理:X射线、r射线、紫外线等
化学:烟焦油、煤等
生物(也就是病毒)
如何避免你就要你要根据自己的情况了1年前查看全部
- illumina测序为什么要消化第二链
狮子只想被爱1年前2
-
asi197977 共回答了22个问题
|采纳率86.4%illumina测序现主要是两端测序,其测序策略为测DNA一条链的前100bp,然后再测两一条链的另一端的100bp,所以在illumina测时候是一条链一条链的测序的.这也是为什么后面要消化掉第二条链的原因.1年前查看全部
- 请看一下这个测序结果是不是缺失了碱基,包括起始密码子和三个碱基
请看一下这个测序结果是不是缺失了碱基,包括起始密码子和三个碱基
GCCGACTGGCCAGCGTTTACTTAAGCTTACCATGGCTTACCCTTACGACGTCCCAGATTACGCGGCTAGGCTGTGCTGCCCACTGGATCCTGCGCGGGACGTCCTTTGTTTACGTCCCGTCGGCGCTGAATCCTGCGGACGACCCTTCTCGGGGTCGCTTGGGACTCTCTCGTCCCCTTCTCCGTCTGCCGTTCCGACCGACCACGGGGCGCACCTCTCTTTACGCGGACTCCCCGTCTGTGCCTTCTCATCTGCCGGACCGTGTGCACTTCGCTTCACCTCTGCACGTCGCATGGAGACCACCGTGAACGCCCACCAAATATTGCCCAAGGTCTTACATAAGAGGACTCTTGGACTCTCAGCAATGTCAACGACCGACCTTGAGGCATACTTCAAAGACTGTTTGTTTAAAGACTAGGAATTCTGCAGATATCCAGCACAGTGGCGGCCGCTCGAGTCTAGAGGGCCCGTTTAAACCCGCTGATCAGCCTCGACTGTGCCTTCTAGTTGCCAGCCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTGACCCTGGAAGGTGCCACTCCCACTGTCCTTTCCTAATAAAATGAGGAAATTGCATCGCATTGTCTGAGTAGGTGTCATTCTATTCTGGGGGGTGGGGTGTTGTATGACAGCTAGGTGGCTTATGGGATTACTTTTTATGTTGTTGTTTTTTCCTGGTTCTTTTGTTTTTTGTCTTTTTTTATTGTTTTTTTTTTTTTTTTTCTTTTTTTTTTTTTTTTTTGCTTTTATTTTTTCTT我想问一下会不会是测序公司的问题还是重组质粒有问题,本人实在是不懂这方面问题.liaoxueliu1年前1 -
铁乐门 共回答了17个问题
|采纳率94.1%测序不是你这样玩的.
待测质粒克隆至少要正向和反向各测一次,如果你对测序公司不放心的话,一个质粒克隆的每个方向要做两个反应,这样每个质粒克隆会得到四个测序结果.比较四个测序结果,方能判断究竟是某个突变是由测序错误造成的,还是确实存在于你的克隆中.你这样没头没脑贴上来一段序列,根本没法帮你.1年前查看全部
- 下一代测序技术有几种
lisaevery1年前1
-
动了谁邀 共回答了20个问题
|采纳率100%按原理分三种:Roche 454 焦磷酸测序,Iiiumina 是边合成边测序,ABI SOLID是 连接测序1年前查看全部
- 我看了你对“转录组测序与转录表达谱测序的异同”的回答我非常感兴趣,
我看了你对“转录组测序与转录表达谱测序的异同”的回答我非常感兴趣,
我现在想做非模式植物的基因表达谱芯片,但是因为没有已知序列,考虑到用EST设计探针做杂交的效果估计不会太好,但用转录组测序后做又有些不太具体了解,不知能否介绍一下转录组测序完成后如何来设计表达谱芯片的探针.jsheal1年前1 -
猫小苗2008 共回答了16个问题
|采纳率81.3%转录组测序后再用RNA-seq做表达谱分析,完全基于测序也可以进行表达谱分析.1年前查看全部
- 如果只有得到的样品中DNA的量很少(DNA是100bp的),拿去测序会有结果吗?
如果只有得到的样品中DNA的量很少(DNA是100bp的),拿去测序会有结果吗?
HPLC能探测到量很少的DNA吗?DNA有带荧光标记.
经HPLC纯化后直接拿去测序可以吗?
要测序的话至少要多少的量呢?
引物序列我知道~
怎么样连到载体上去呢?HPLC纯化后要先PCR在连吗?是不是要做个克隆了?
如果量不够的话,HPLC后PCR下,拿PCR的产物测序可以吗?
再谢再谢!fovai1年前2 -
高山长风 共回答了20个问题
|采纳率85%100bp的片段去直接测序很困难.并且你有该片段两侧的引物序列么?
建议你还是做一步简单的连接反应将该片段链接到载体上去测序,这样比较保险些.
测序时DNA在溶液中的浓度最好大于100ng/微升.如果提供10微升样品测序的话,也就是需要1微克的DNA.
是要做个克隆.1年前查看全部
- 果蝇作为实验材料,被广泛应用于遗传学研究的各个方面.果蝇也是早于人类基因组计划之前其基因被测序的一种生物.请据如图回答:
果蝇作为实验材料,被广泛应用于遗传学研究的各个方面.果蝇也是早于人类基因组计划之前其基因被测序的一种生物.请据如图回答:

(1)对果蝇的基因组进行研究,应测序______条染色体,它们是______
A.3条常染色体+XB.3条常染色体+Y
C.3对常染色体+XYD.3条常染色体+XY
(2)假如果蝇的红眼(E)和白眼基因(e)位子X染色体上,图中的甲表示某果蝇个体的减数分裂,则:
①图中Ⅱ、Ⅲ细胞的名称分别是______细胞、______细胞
②若不考虑交叉互换,图中I细胞通过减数分裂可产生______种类型的配子.
③若图甲果蝇与白眼果蝇杂交,后代中有红眼果蝇,则图甲果蝇的眼色为______色;
该果蝇减数分裂产生的细胞a与一只红眼果蝇产生的配子结合并发育为一只白眼果蝇,则这只白眼果蝇的性别为______;请在图乙中标出I细胞相关的眼色基因,并在图丙中画出I细胞产生的细胞c的染色体组成.shui_lian1年前1 -
sunshineliao 共回答了23个问题
|采纳率91.3%解题思路:果蝇因具有易饲养、繁殖周期短、具有易区分的相对性状、遗传组成相对简单等优点而常被用作遗传学的实验材料.其细胞内含有4对8条染色体,雄性个体为6+XY,雌性个体为6+XX,由于X与Y染色体的基因组成不同,所以在对果蝇基因组进行研究,应测序3对常染色体各一条以及性染色体X与Y共5条染色体.果蝇的红眼和白眼是一对相对性状,若基因位于常染色体上,则红眼基因型为EE和Ee,白眼基因型为ee;若基因位于X染色体上,则红眼基因型为XEXE、XEXe和XEY,白眼基因型为XeXe和XeY.XY为一对同源染色体,在减数分裂过程中,同源染色体分离,分别进入不同的配子中去.(1)果蝇含有4对同源染色体,最后一对是性染色体(XX或XY),所以对果蝇基因组进行研究时,应测序5条染色体,即3+XY.
(2)①由于图甲表示某果蝇个体的减数分裂,所以图中Ⅰ、Ⅱ、Ⅲ细胞的名称分别是精原细胞、次级精母细胞和精细胞.
②若不考虑交叉互换,图中I细胞通过减数分裂可产生含X染色体和含Y染色体的2种类型的配子.
③若图甲果蝇与白眼果蝇杂交,后代中有红眼果蝇,则图甲果蝇基因型为XEY,其眼色为红色,该果蝇减数分裂产生的细胞a与一只红眼果蝇产生的配子结合并发育成一只白眼果蝇,说明细胞a中不含XE,所以含有Y染色体,则这只白眼果蝇的性别是雄性.
故答案为:
(1)5 D
(2)①次级精母 精 ②2 ③红 雄性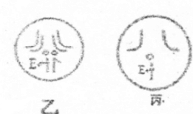
点评:
本题考点: 细胞的减数分裂.
考点点评: 本题以果蝇染色体组成为背景,考查减数分裂的相关知识,意在考查学生的识图能力和判断能力,运用所学知识综合分析问题和解决问题的能力.1年前查看全部

- 如何知道NCBI中某基因组的发表时间(或测序时间)
长袖善舞09031年前1
-
宁波26 共回答了22个问题
|采纳率81.8%一般上面的时间是序列提交到NCBI上面的时间
比如你搜索Escherichia coli吧
在genomes里面会出现:
1 Genome:genome sequencing projects by organism
点1进去,出现:
Reference genomes:[see all organisms]
Escherichia coli str.K-12 substr.MG1655
Escherichia coli O157:H7 str.Sakai
Escherichia coli IAI39
Escherichia coli UMN026
Escherichia coli O83:H1 str.NRG 857C
Escherichia coli O104:H4 str.2011C-3493
点开第一个,找到RefSeq下面的NC_000913.2:
Type Name RefSeq INSDC Size (Mb) GC% Protein rRNA tRNA Other RNA Gene Pseudogene
Chr - NC_000913.2 U00096.2 4.64 50.8 4,145 22 89 65 4,497 181
再进去,找到COMMENT,里面一般会说是什么时候提交的,如下:
COMMENT REVIEWED REFSEQ:This record has been curated by NCBI staff.The
reference sequence is identical to U00096.
On Jun 24,2004 this sequence version replaced gi:16127994.1年前查看全部
- 我最近读了篇文献是关于基因组再测序(Whole-genome re-sequencing),但对其还不是很了解,麻烦介绍
我最近读了篇文献是关于基因组再测序(Whole-genome re-sequencing),但对其还不是很了解,麻烦介绍一下流离失所1年前1
-
huatuoa 共回答了27个问题
|采纳率85.2%全基因组再测序
尽管同胞配-配对和全基因组相关研究可以鉴定对复杂疾病有主要作用的基因和普通疾病相关的等位基因,但是对于罕见的等位基因用这种方法还是不能检测到.罕见的等位基因只有通过突变扫描或基因再测序才能发现.目前已经在寻找疾病的候选基因时使用上述方法.但是全基因组方法在鉴定无论隐性还是显性作用的等位基因方面都将更为有效.将这种方法合理地推广到全基因组重复测序.例如,假如对50-100例某种疾病的患者进行基因组再测序,可以在这些个体中发现影响该病的大部分变异基因.如果在某一特定的基因中发现罕见的、严重的等位基因,就可以在更多的病人中详细研究这个基因(同胞-配对或病人-对照).类似地,如果一个SNP在普通人群中是罕见的,但在疾病组中频发,那么这个SNP就可能在更大量的病人中被鉴定并证实真的与疾病相关.当然这种方法也会发现许多可能与疾病无关的变异基因.但是,当基因型测序的费用下降时,这些基因变异就有可能在更多的样本数据库中得到评价和鉴别.基因组测序所使用的大片段(~50Kb)插入克隆也可以用来鉴定插入、复制、缺失和倒转的变异.通过将这些克隆末端序列与参考序列比对,将发现缺失和插入分别表现为序列更加靠近或远离.复制和倒转将表现为克隆末端的序列方向错误.如果将这些方法的应用范围扩大,通过芯片检测克隆的末端序列,将可实现整个基因组的扫描.1年前查看全部
- 这是细菌16S PCR产物电泳图像,大小为1.5kb,但条带上下出现弥散现象,测序波峰出现重峰现象
海波00001年前4
-
千分之91思远 共回答了12个问题
|采纳率83.3%细菌16S rRNA基因序列是集高度保守和变异性与一身的,现在采用的多数PCR扩增引物都是采用的所有细菌的通用引物,即:几乎能够扩增出所有的真细菌.所以,16S rDNA的扩增面临的主要问题是污染,只要有其他细菌DNA的污染,就可能出现假阳性或测序重叠峰,也就是说一条1500bp的带可能是多个细菌的16s rDNA的扩增产物的混合物.
解决这一问题需要注意的事项很多:
1.所有的物品和试剂都要严格防污染,包括taq酶,有很多公司产的聚合酶是用细菌生产的,如果处理纯化不当可能含有细菌的DNA;
2.扩增模板开始时一定要选取单个菌落来制备;
3.由于该基因在多数细菌基因组内是多拷贝的,所以模板DNA的量不宜太多,否则有非特异性扩增(你的带看上去模板DNA就有些过量)
4.测序宜采用连接T载体后进行,不宜采用PCR产物直接测序的方法.
5.每次PCR都要设立未加模板的空白对照!
6.照片中引物二聚体也很多,需适当减少反应体系中引物的用量.1年前查看全部
- 关于通用测序引物的问题现在要克隆一段基因,设计引物时,上游加入HindⅢ酶切位点,下游加入KpnI酶切位点.师兄要求再设
关于通用测序引物的问题
现在要克隆一段基因,设计引物时,上游加入HindⅢ酶切位点,下游加入KpnI酶切位点.师兄要求再设计一段通用测序引物,用来检测质粒重组时,目的基因片段是否连在质粒上.
那么这个通用测序引物,该怎样设计?是不是针对于HindⅢ和KpnI这两个酶切位点?波菜叶子1年前1 -
五饼贰鱼 共回答了19个问题
|采纳率89.5%用质粒上的序列设计引物.不要跨越这两个酶切位点.就是在插入位点的上下游设计引物.这样才是“通用引物”,可以用于所有使用该质粒载体的PCR检测.1年前查看全部
- 耗时13年,花费4.37这是人累彻底完成自身基因组测序所付出的时间和金钱.下面关于DNA描述,错误的是:
耗时13年,花费4.37这是人累彻底完成自身基因组测序所付出的时间和金钱.下面关于DNA描述,错误的是:
A.DNA是双螺旋结构,而且黏性极强
B.DNA长达2米,能够困住微生物
C.DNA是遗传信息的载体,无妻xx1年前5 -
sdvjpf6 共回答了16个问题
|采纳率100%C1年前查看全部
- 谁知道microRNA芯片、microRNA测序和基因表达谱芯片区别,具体些,
pocky52521年前1
-
该i飞机 共回答了25个问题
|采纳率96%microRNA芯片和基因表达谱芯片都是芯片.
芯片指样本荧光染色杂交到芯片上,通过各个点亮度差异来判断表达差异的方法.
区别是一个针对microRNA,微小RNA.另一个针对mRNA,信使RNA.
microRNA测序区别于芯片分析方法.直接测出小片段的序列然后用生物统计方法合并区段等最终计算出表达差异.
microRNA芯片与microRNA测序是用不同平台研究同样的问题.1年前查看全部
- 测序结果对分子生物学研究的意义,列举几种基因组测序结果产生的最重要的意义
king577771年前2
-
bc600lwp 共回答了21个问题
|采纳率85.7%1发现新基因,确定基因产物的功能,进而运用这些信息研究遗传与人的健康与疾病的关系.
为各种遗传病的诊断、治疗及新药开发提供了分子基础.如目前在遗传病的诊断方面,单基因疾病的检测方法有单链构象分析法( single strand conformation)、酶错配切断法(Enzyme mismatch cleavage)、蛋白质截断法(protein truncation test).对于多基因疾病,尽管目前还没有很完善的诊断方法,但已经成功的将一些可疑基因定位在染色体6的HLA区,这个区段有200多个基因,已经鉴定有一百多种疾病与这个区域有关系.人们都希望通过基因治疗彻底的治愈遗传病,然而,目前在基因治疗取得的成果极其有限,只有体外转移基因到骨髓细胞应用于临床.不过,人类基因工程将加快基因的发现,提高疾病的检测手段.斯坦福大学已经建立了60种癌细胞中8000多种基因的表达谱,通过比较癌变细胞和正常细胞基因表达差异,将能很快的确定治病机理,提供治疗所必需的信息.在新药开发方面,生物信息学工具和数据库可用于鉴定新基因,寻找药物作用的靶位点.
基因组学的发展,还大大加快了农作物育种.如从分子生物学水平上鉴定与农作物形状相关的基因,现在有基因组测序、运用 ESTs序列和基因芯片技术,运用生物信息学方法搜寻,运用蛋白质组学、功能基因组学等多种方法.一旦发现有助于提高农作物产量的基因,可应用先进的生物技术,将其介导进农作物中,改良农作物的遗传性状.目前,运用Bt转基因系统转入抗疾病基因得到的转基因作物有玉米、棉花,土豆.另外,已将维生素A 合成系统相关的基因转入进水稻中,可得到含维生素A的大米,这样就可以解决东南亚儿童由于缺乏维生素A引起的失明问题.1年前查看全部
- 为什么基因测序前将基因PCR克隆后还要放到大肠杆菌载体上再测序?
为什么基因测序前将基因PCR克隆后还要放到大肠杆菌载体上再测序?
除了防止PCR产物降解外还有别的原因吗?fantasysky1年前3 -
淝水之战 共回答了18个问题
|采纳率66.7%一楼完全没说到重点,二楼正解.
你看过测序的结果就知道了,前面的几十个碱基是无法识别的杂峰,最后几百个碱基也是无法识别的杂峰,尽管结果中还是以单个碱基字母的形式给你写出来,但是那是0智商的机器自动记录的,你要人工剔除这部分序列,也就是说你不知道这部分序列的真实数据.
放到载体上就是为了把前面这几十个测序弄不出来的碱基放到质粒的部分,不清楚就不清楚,我们反正不需要知道,我们需要知道的序列完全落在后面清楚的部分就行了.1年前查看全部
- 细菌16s rdna测序后怎么比对和构建系统发育树,老师不管我们了
box41年前1
-
zj2677 共回答了18个问题
|采纳率88.9%有很多方法,可以下载软件也可以在线构建.
在线构建可将你的序列输入PUBMED的Blastn中进行对比,结果中有一种就是系统发育树,但里面残余对比的序列太多,逆序挑选一下.
软件有很多,比较简单的是DNAstar中的Megalin,但结果比较粗.
推荐用Clustal X软件进行同源性比对后,运用MEGA3.0软件进行系统发育分析并绘制系统发育树,也很简单,看看说明就会.
比较公认的严谨的是philip系列软件,但操作复杂.1年前查看全部
- 菌液PCR的时候能P出目的条带,为什么测序却完全不正确?
rechelwong1年前1
-
newff22 共回答了17个问题
|采纳率88.2%菌落PCR的假阳性高很可能是因为你做PCR的循环数过大,我们实验室一般对于高拷贝数的质粒,循环数不会超过25个,一般用25个.假阳性出现的原因关键在于做连接的时候加了比较多的目的基因的酶切产物,在转化时,那些没有连接入载体的PCR产物,有的会沾在感受态细胞上,还有的虽然在溶液中,涂板时一起就一起涂在板上了,挑菌的时候可能会挑到,即使没挑到,但菌表面的基因片段也有可能被扩增,如果你的循环数太高,就会在PCR时呈现阳性结果.希望能帮到你1年前查看全部
- 请问测序图谱中自始至终都是套峰,且峰图不乱一高峰里套一矮峰是怎么回事?
王峰741年前1
-
法行动 共回答了16个问题
|采纳率68.8%产生套峰原因:假若为菌或者质粒为双位(引物结合位点不单一)建议换引物测序实验,另外一个是由于存在污染重新挑单克隆进行测序实验即可.
若为PCR一个是由于存在非特异扩增.一个是引物特异性不好.另外一个原因是引物为兼并引物.'
假若套峰小峰为前或后的主峰说明是由于移码造成既引物合成存在问题.可以把测序结果发给合成公司重新合成.
以上供参考!具体原因请发峰图参考!1年前查看全部
- 问个很菜的问题:在跑完pcr以后目的基因已经扩增了很多了,然后再切胶纯化后应该就可以用来去测序了,为什么还要进行后面的连
问个很菜的问题:在跑完pcr以后目的基因已经扩增了很多了,然后再切胶纯化后应该就可以用来去测序了,为什么还要进行后面的连接转化重组质粒的抽提和酶切鉴定啊,他们有什么意义?wenge3211年前2
-
skycrazy 共回答了18个问题
|采纳率83.3%你胶回收回来后,虽然看上去用来测序的量是够了,但是你的基因总不是只用来测个序吧,还有很多的测序工作需要完成,比如基因表达、保种等等.将基因连接上质粒后,转入细胞内,可以根据不同载体的功能来研究该基因,或者扩增该基因以及带基因的载体.可以将质粒抽出来保种和菌液保种,一切都要给自己留个后路和实验成果不是~酶切鉴定就只是个鉴定而已,看看基因是否连在了载体上.1年前查看全部
- 基因组计划的DNA测序原理
zhoutian19851年前1
-
草莓的幸福 共回答了10个问题
|采纳率90%双脱氧终止法.在测序用的缓冲液中含有四种dNTP及聚合酶.测序时分成四个反应,每个反应除上述成分外分别加入2,3-双脱氧的A,C,G,T核苷三磷酸(称为ddATP,ddCTP,ddGTP,ddTTP),然后进行聚合反应.在第一个反应物中,ddATP会随机地代替dATP参加反应一旦ddATP加入了新合成的DNA链,由于其3位的羟基变成了氢,所以不能继续延伸.所以第一个反应中所产生的DNA链都是到A就终止了; 同理第二个反应产生的都是以C结尾的; 第三个反应的都以G结尾,第四个反应的都以T结尾,电泳后就可以读出序列了.也许这样说你不一定明白.举一个例子,假如有一个DNA,互补序列是GATCCGAT,我们试着做一下:
在第一个反应中由于含有dNTP+ddATP,所以遇到G,T,C三个碱基时没什么问题,但遇到A时,掺入的可能是dATP或ddATP,比如已合成到G,下一个如果参与反应的是ddATP则终止,产生一个仅有2个核苷酸的序列:GA,否则继续延伸,可以产生序列GATCCG,又到了下一个A了.同样有两种情况,如果是ddATP掺入,则产生的序列是GATCCGA,延伸终止,否则可以继续延伸,产生GATCCGAT.
所以在第一个反应系统中产生的都是以A结尾的片段:
GA,GATCCGA,
同理在第二个反应中产生的都是以C结尾的片段:
GATC,GATCC,
在第三个反应中产生的都是以G结尾的片段:
G,GATCCG
在第四个反应中产生的都是以T结尾的片段:
GAT,GATCCGAT,
电泳时按分子量大小排列,A反应的片段长度为2,7; C反应的为4,5; G反应的为1,6; T反应的为3,8,四个反应的产物分别电泳,结果为
8 7 6 5 4 3 2 1
A | |
C | |
G | |
T | |
我们可以从右向左读,为GATCCGAT,至此,测序完成(上面这个图在百度知道中显示不正常,因为百度知道的网页用的是比例字体,你如果想看它,拷贝到记事本中,用等宽的字体来看).1年前查看全部
- 如何用全基因外显子测序家族突变基因
宇智波雷翼1年前1
-
原始森林种植者 共回答了9个问题
|采纳率66.7%如果研究家族的遗传信息的话外显子组测序获得所有变异,主要是SNP和indel这种小型的突变(序列上来说涉及的bp数比较少)根据遗传信息,判别突变类型,进行所有变异的筛除.1年前查看全部
- 在DNA测序工作中,需要将某些限制性内切酶的限制位点在DNA上定位,使其成为DNA分子中的物理参照点.这项工作叫做“限制
在DNA测序工作中,需要将某些限制性内切酶的限制位点在DNA上定位,使其成为DNA分子中的物理参照点.这项工作叫做“限制酶图谱的构建”.假设有以下一项实验:用限制酶HindⅢ,BamH I和二者的混合物分别降解一个4kb(1kb即1千个碱基对)大小的线性DNA分子,降解产物分别进行凝胶电泳,在电场的作用下,降解产物分开,如图所示.
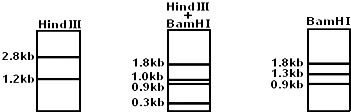
据此分析,这两种限制性内切酶在该DNA分子上的限制位点数目是以下哪一组?( )
A. HindⅢ1个,BamHⅠ2个
B. HindⅢ2个,BamHⅠ3个
C. HindⅢ2个,BamHⅠ1个
D. HindⅢ和BamHⅠ各有2个27631年前1 -
给你快乐 共回答了19个问题
|采纳率89.5%解题思路:限制性核酸内切酶(限制酶)具有特异性:能够识别双链DNA分子的某种特定的核苷酸序列,并且使每一条链中特定部位的两个核苷酸之间的磷酸二酯键断开.根据题中图示,HindⅢ将该DNA分子切割成2种片段,说明有1个切点;BamHⅠ将该DNA分子切割成3种片段,说明有2个切点.根据题中图示,可将4kb的DNA示意图画出,再在此DNA上将两种限制酶及二者的混合物分别降解结果的示意图画出,进行比较,即可推出HindⅢ的切点为1个,将DNA切成2.8kb和1.2kb两段,同理BamHⅠ有2个,分别在距DNA一端的0.9kb处和另一端的1.8kb处.
故选:A.点评:
本题考点: 基因工程的原理及技术.
考点点评: 本题考查了限制性核酸内切酶的相关内容,意在考查考生能理解所学知识的要点,把握知识间的内在联系,能用数学方式准确地描述生物学方面的内容、以及数据处理能力.1年前查看全部
- 测序通量和测序读长是什么意思急,急
lpoaiw1年前1
-
逸游之星 共回答了13个问题
|采纳率92.3%没有测序通量这个说法.我觉得您可能是看到了“高通量测序”这个词,这个词是相对于旧时的桑格尔测序而言的,因为测序量大,一次并行对几十万到几百万条DNA分子进行序列测定,所以被称为高通量测序.
而测序读长是说测序得到的读段(reads)长度.一般测序的过程是:实验室提取DNA(RNA)->送公司测序.在测序中,是把我们提取好的样品DNA随机打成小片段进行的,这些小片段就被称为reads,他们的长度就是读长.1年前查看全部
- 重组质粒后为什么还需要双酶切、PCR鉴定并测序分析
duhaha1年前1
-
强调的工 共回答了27个问题
|采纳率92.6%因为你的重组质粒不知是否构建成功,通过双酶切,PCR看是否连上去,且方向是否正确,
好好把书看看吧,这样有了完善的理论,做起实验来才明白的透彻,而不是盲目的,也能自己分析!1年前查看全部
- 连接载体如何测序?想知道做实验如何操作?用pGM-T载体可以做真菌测序吗?谢谢
22175661年前1
-
喜世 共回答了15个问题
|采纳率93.3%如果你重组的载体在多克隆两边有通用引物的话直接用通用引物测序就行了,没有的话得叫测序公司帮你合成.你是要测真菌的什么东西,是基因组,还是它的某一个基因?你只要能够把你要测序的那段基因重组到这个T载体上就可以测序的.1年前查看全部
- 测序胶电泳的分辨水平是A.10个碱基 B.1个碱基 C.100个碱基 D.5个碱基
车之友广州学车1年前1
-
老式枪 共回答了19个问题
|采纳率84.2%C.100个碱基1年前查看全部
- 什么是普通的基因测序,它和高通量测序有什么区别吗?
zxzxzhuxu11年前3
-
灌水专用 共回答了25个问题
|采纳率96%“普通的基因测序”应该是指“常规DNA测序”吧,是用Sanger法(也就是双脱氧法)进行测序的方法,目前非常普遍的是直接用ABI 3730xl 进行的自动测序,基本上可以做到600bp-800bp的读长.
高通量测序的概念其实是一个相对的概念,在2000年的时候,3700、MegaBace等仪器上的测序也是高通量测序,是相对手工测序或者跑平板胶来说的.
不过到2005年以后,高通量测序就改指第二代测序(Next generation sequencing),454、Solexa(后改为Illumina)和SOLiD等第二代测序,比3730等第一代测序的通量提高了成千上万倍,甚至上亿倍,所以称为高通量测序.
NGS的特点主要有:
1、通量高,一个RUN能产生500Mb-600Gb的数据量
2、读长相对较短,454(约400-500bp),Illumina(100-250bp),SOLiD(75-100)
3、单位数据的成本非常低,现在很多项目测序的费用,已经非常低,生物信息分析成本变得更为重要了.1年前查看全部
- 为什么待测的DNA分子首先要解旋变为单链,才可用基因探针测序?
痞子瞌睡虫1年前2
-
依恋你 共回答了16个问题
|采纳率93.8%因为基因探针(一般来说)就是与DNA的其中一条链特异结合形成双螺旋结构才稳定的啊.待测DNA不解旋的话探针就结合不上去.1年前查看全部
- (2003•上海)“人类基因组计划”中的基因测序工作是指测定( )
(2003•上海)“人类基因组计划”中的基因测序工作是指测定( )
A.DNA的碱基对排列顺序
B.mRNA的碱基排列顺序
C.蛋白质的氨基酸排列顺序
D.DNA的基因排列顺序最怕想名字1年前1 -
游哉乎悠哉也 共回答了20个问题
|采纳率90%解题思路:人类基因组计划是指完成人体24条染色体上的全部基因的遗传作图、物理作图、和全部碱基的序列测定.通过该计划可以清楚的认识人类基因的组成、结构、功能极其相互关系,对于人类疾病的诊治和预防具有重要的意义.根据人类基因组计划的定义可知,基因测序工作是指测定基因中DNA的碱基对排列顺序.
故选:A.点评:
本题考点: 人类基因组计划及其意义.
考点点评: 本题比较简单,考查了人类基因组计划的基因测序工作,该考点考生识记即可,意在考查考生的识记能力和理解能力.1年前查看全部
- 关于基因测序技术Illumina Genome Analyzer测序工作原理:边合成边测序.即生成新DNA互补链时,要么
关于基因测序技术
Illumina Genome Analyzer测序工作原理:边合成边测序.即生成新DNA互补链时,要么加入dNTP通过酶促级联反应催化底物激发出荧光,要么直接加入被荧光标记的dNTP或半简并引物,在合成或连接生成互补链时,释放出荧光信号.通过捕获光信号并转化为一个测序峰值,获得互补链序列信息.
上面那段话中最后那句是什么意思?
是不是测序峰值的高低代表不同dNTP发出的荧光信号?aokyb1年前1 -
我知我傻的可以 共回答了13个问题
|采纳率100%上面这段话是普通二代测序(454测序法)的原理,不同的荧光信号表示不同的碱基.
你说的没错,测序结果返还给客户的时候一般都有两个文件,一个是Seq,一个是ab1,其中ab1这个文件就是测序峰图,测序结果就是根据峰图确定的.你也可以自己尝试检查这个峰图,来确定有些检测是不是测出来了,但是转换错了.
有问题我们可以继续交流.1年前查看全部
- 你有在做基因组测序?目前在做人类一个癌症基因样本的测序,希望人类基因组研究或者测序过的人交流下reads。
fan20061年前3
-
珊羽舒 共回答了25个问题
|采纳率88%没有.1年前查看全部
- 点突变后蛋白质功能不变分离出一个基因的mRNA,转录获得cDNA,测序后得知该基因发生了点突变,但该基因表达的蛋白功能没
点突变后蛋白质功能不变
分离出一个基因的mRNA,转录获得cDNA,测序后得知该基因发生了点突变,但该基因表达的蛋白功能没有发生改变,为什么?e度恋情1年前2 -
网球好男儿 共回答了25个问题
|采纳率100%可能有两个原因:
1.因为密码子具有简并性,即大部分氨基酸都有2个或2个以上的密码子,基因虽然发生了点突变,但很有可能突变成编码同一氨基酸的其它密码子,翻译出来的蛋白质氨基酸序列不变.
2.基因发生点突变,使得对应的密码子变成了编码另一个氨基酸的密码子(一般是性质相似的氨基酸),然而原来的氨基酸对于蛋白质的功能来说,并没有很大的影响,这时候蛋白质功能基本保持不变.1年前查看全部
- 引物扩增片段太小(360bp),跑普通PCR去克隆测序可行不
咖啡不加方糖1年前4
-
失忆的柏拉图 共回答了13个问题
|采纳率69.2%看你是实验目的是什么:
如果是为了对目的基因片段测序,用普通taq酶pcr没问题,而且,也没必要做到克隆里再测,可直接用pcr产物测;
如果是为了做克隆已备后续实验,建议你用高保真的taq酶,普通taq错的随时随地,运气好一次就成了,运气不好,能把人折磨死.1年前查看全部
- GelRed染色是否会对后续的TA克隆、测序、酶切产生影响?
GelRed染色是否会对后续的TA克隆、测序、酶切产生影响?
因实验室限制使用EB,本人采用核酸染料GelRed染色后,切胶回收PCR产物,然后采用Takara pmd 19-T simple vector试剂盒连接,严格按照说明书操作,感受态细胞为JM109(商品化).蓝白斑筛选结果阴性对照没有菌落,阳性对照(试剂盒自带pUC19)均为白色菌落,但是实验平板上却全是蓝色菌落,不知道是否是由于采用GelRed染料原因,对TA克隆造成影响?求各位大侠赐教!GelRed染色是否会对后续的克隆、测序、酶切产生影响?什么是ll1年前2 -
天天4017 共回答了17个问题
|采纳率94.1%我们实验室就是用GelRed染色,对后续试验没有产生过影响.
切胶回收后,理论上是没有GelRed了,不会对后续试验产生影响.1年前查看全部
- PCR-DGGE检测厌氧反应器中的微生物,为何测序回来的结果大都是所谓的宏基因组呢?与预期的一点边都不沾啊
PCR-DGGE检测厌氧反应器中的微生物,为何测序回来的结果大都是所谓的宏基因组呢?与预期的一点边都不沾啊
“Metagenome sequence ctg1167,whole genome shotgun sequence” 宏基因组序列ctg1167,全基因组鸟枪序列“Marine metagenome ”海洋宏基因组“Compost metagenome contig”宏基因组堆肥重叠群"Human gut metagenome DNA"人类肠道宏基因组DNA------这些跟我的反应器中的微生物几乎没有关系啊,正常应该是厌氧氨氧化菌、产甲烷菌、变形菌等等的啊,郁闷啊,是我做的有问题,还是测序有问题,还是反应器就这样,还是别的,要交毕业论文了,这些还没弄明白,着急,大师来了1年前1 -
梦峡谷 共回答了15个问题
|采纳率86.7%话说我也在说毕设.lz你做的是厌氧反应器中的微生物,那么你测的是16s V3区的pcr 还是整个16s的pcr.PCR-DGGE之后你是直接割胶纯化测序,还是连质粒转化然后测序的啊.求你做的顺序,我要作参考.1年前查看全部
- 生物问题24.11下列表述正确的是( )A.先天性心脏病都是遗传病B.人类基因组测序是测定人的23条染色体的DNA碱基
生物问题24.11
下列表述正确的是( )
A.先天性心脏病都是遗传病
B.人类基因组测序是测定人的23条染色体的DNA碱基序列
C.通过人类基因组计划的实施,人类可以清晰地认识到人类基因的组成、结构和功能
D.单基因遗传病是由一个致病基因引起的遗传病
fqfm32542901年前1 -
Emoda 共回答了20个问题
|采纳率85%C [解析] 先天性心脏病目前来说主要与母亲孕期病毒感染、接触放射线等有关,而与遗传关系不大,故A项错误。人类基因组测序是测定人的22条常染色体及X、Y 2条性染色体上DNA的碱基序列,故B项错误。通过人类基因组测序,可以认识到基因的组成、结构和功能,故C项正确。单基因遗传病是由一对等位基因控制的遗传病,故D项错误。1年前查看全部
- 液氮速冻,干冰保存的纯化单菌落拿到公司做重测序,需活化后提DNA还是直接不经过活化,融化后直接提DNA?
xwrcqx1年前2
-
jswxck 共回答了22个问题
|采纳率100%建议先活化后再提DNA,不然的话提出来的DNA浓度也很低1年前查看全部
- 什么是pair-end 测序?只测一个片段的两端吗,那中间的序列怎么得知?有什么优点?
什么是pair-end 测序?只测一个片段的两端吗,那中间的序列怎么得知?有什么优点?
如题,谢谢大家了.elain19781年前1 -
lemon_tree28 共回答了19个问题
|采纳率94.7%Paired-end方法是指在构建待测DNA文库时在“两端”的接头上都加上测序引物结合位点,在第一轮完成后,去除第一轮测序的模板链,用对读测序模块引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量
优点:
pair end是直接在DNA两端假设接头进行双向测序,插入片段长度较短1年前查看全部
- 人类基因组计划中的基因测序是指DNA的基因排列顺序还是DNA上的所有碱基对的排列顺序?
qgxlxm1年前5
-
bingyuangel 共回答了20个问题
|采纳率95%指的是DNA上的所有碱基对的排列顺序1年前查看全部
- 为什么人类基因组计划测序只用测24条染色体
为什么人类基因组计划测序只用测24条染色体
一对同源染色体上的DNA序列不同,哪46条染色体的DNA序列应该都不同啊.应该全部测啊wesleyying1年前3 -
苍山血绿 共回答了19个问题
|采纳率100%人类是22+XY对染色体,除XY外,其他22对的每对染色体的两条序列都是一样的,所以只要测其中一条序列就可以了.1年前查看全部
- PCR产物纯化后,连接载体PGMT,转化如DH5α后为何不能直接测序,还要再酶切和提取质粒呢?
彩怡1年前2
-
乐乐晓蒙 共回答了21个问题
|采纳率90.5%你们是自己测序,还是送公司测序?
如果是送公司的话,可以直接测序,测序引物为M13通用引物.但是测序之前需要把挑出来的克隆,扩繁、PCR检测后再测序.测序公司再提取质粒-测序.
如果是自己测序,酶切应该是检测用,阳性的质粒再测序.
总之,一般情况下,测序都是用质粒.酶切或者PCR只是用来检测阳性克隆,酶切更准确.现在PCR产物也可以直接用来测序.1年前查看全部
- 我做的是双向测序,片段300bp左右.请问怎么在测序结果中找出引物?需不需要把某个片段或序列变方向之类的
zhengtong781年前2
-
茉美眉 共回答了15个问题
|采纳率80%正向测序结果可以找到反向引物的反向互补序列
反向测序结果可以找到正向引物的反向互补序列1年前查看全部
- PCR产物测序需要测整个片段吗?
PCR产物测序需要测整个片段吗?
PCR产物要看一下是不是目的片段,需要将扩增出来的整个片段测序吗?具体应该怎样做?尚玄乐2971年前3 -
魔恋仙 共回答了21个问题
|采纳率81%最好把pcr产物测通,这样最准确.
具体的很简单,直接把pcr产物给测序公司,就可以了.要求是条带要单一,含量要足够.
可以咨询相关的测序公司.1年前查看全部
- 5'race 只扩增出一条带 测序回来不对,能找到通用引物,找不到特异引物,是什么原因啊
spcflying1年前1
-
jingxingweixian 共回答了23个问题
|采纳率95.7%可以只加入通用引物做一个control,怀疑你扩出来的片段是只有通用引物结合扩增出来的,而特异性引物并没有结合1年前查看全部
- RPKM是衡量测序数据的什么值?
寓身化世一尘沙1年前1
-
tt八股文 共回答了15个问题
|采纳率80%mapping结果的情况.
reads per kilobase of exon model per million mapped reads1年前查看全部
大家在问
- 1单项选择 He was born ( )Beijing ( )1993.A.on,inB.in,inC.at,onD.o
- 26.97乘以99+6.97的简便运算是?
- 3today is sunday,we don't go to school
- 4新制氯水具有的性质(表格)参与反应粒子:CL- H+ HClO Cl2所加试剂:() () () ()实验现象:() (
- 5一定温度下,在2L的密闭容器中,X.Y.Z三种气体的物质的量随时间变化的曲线如右图所示:该反应的方程式为
- 6用四个成语造一段 记住,是一段话 100字左右 还要有某本书中的段落缩写(原段也要写,并写出出自哪本书
- 7某班学生人数在40人到50人之间,男生和女生人数的比是5:6,这个班有男生______人,女生______人.
- 8英语翻译:人们说蛇是危险的动物
- 9选择正确的单词。 ( ) 1. 在那时
- 10圆的直径是10,一张长方形的纸,剪下图中的一个圆及长方形,正好做成一个无盖的圆柱,求圆柱的体积.
- 11英语翻译还是翻成:畅享与众不同的飞行风格
- 12连词成句dessert,for,has,ice cream,brother,hishealthy,running,foo
- 13Lim 〔1+a+a^2+a^3+…….+a^n〕/〔1+b+b^2+b^3+…+b^n〕帮我算一下这道题,我需要整个运
- 14人的血糖一天内是怎样变化的
- 15下面的几组线段中,不能组成三角形的有( ).